Inhibition der Glutaminolyse und Glycolyse zur Hemmung der C. parvum-Infektion sowie One Health-Studie zur Kryptosporidiose in Kamerun
Projektbeschreibung
Kryptosporidien sind parasitäre Durchfallerreger beim Menschen, die insbesondere in Entwicklungsländern bei Kleinkindern und HIV-Infizierten zu hohen Morbiditäten und Todesfällen führen. Dabei sind die genauen infektionsbedingten Zusammenhänge in diversen Entwicklungsländern, wie z. B. Kamerun, nicht vollständig untersucht. Zur Therapie der Risikogruppen sind derzeit keine effektiven Medikamente erhältlich. Kryptosporidien sind obligat intrazelluläre Protozoen und verfügen selbst nur über minimale eigene Stoffwechselkapazitäten, daher müssen sie den Stoffwechsel der Wirtszelle zu ihrem Vorteil modulieren, um sich intrazellulär zu vermehren. Über die Charakterisierung der metabolischen Signaturen C. parvum-infizierter Wirtszellen konnten wir Stoffwechselreaktionen und -wege der Wirtzelle identifizieren, die für die Parasitenvermehrung essenziell sind.

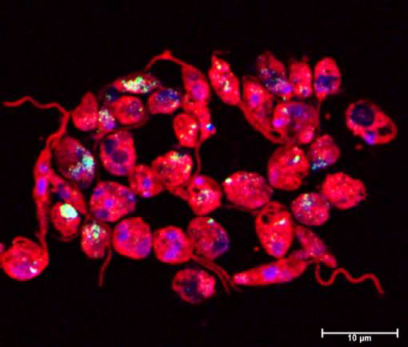
Cryptosporidium parvum- (gelb) infizierte Wirtszellen (Zellkerne: blau), tomografische Mikroskopie © Juan Vélez
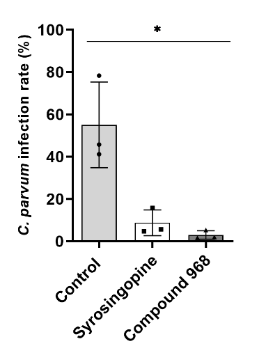
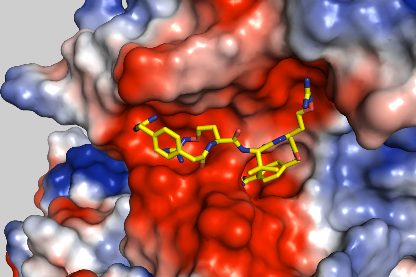
Hemmung von Cryptosporidium parvum über Stoffwechselinhibitoren (Vélez et al. 2021c)
Wissenschaftliches Ziel:
Das Projekt zielt darauf ab, strategische Angriffspunkte im Stoffwechsel der Wirtszelle (v. a. Glykolyse, Glutaminolyse) über neue Inhibitoren oder Kombinationsbehandlungen zu blockieren und damit indirekt die Parasitenentwicklung zu hemmen. Daneben wird eine One-Health-Studie zur Kryptosporidiose in Kamerun unter Berücksichtigung diverser epidemiologisch wichtiger Parameter durchgeführt.
DRUID-Kooperationspartner:
E4 Spengler lab
Literatur D4: 1. *Vélez et al. (2021a) Pathogens 11(1):49. 2. * Vélez et al. (2021b) Biology 10(10):963 3. ** Vélez et al. (2021c) Biology 10(1):60