Projektbereich A:
A1Interaktionen von Ebola-Virus-Proteinen als Ziele antiviraler Strategien
Stephan Becker

Prof. Dr. Stephan Becker
Institut für Virologie
Philipps-Universität Marburg
Hans-Meerwein-Str. 2
35043 Marburg
Phone: +49 (0)6421-28 66253
E-Mail: becker(at)staff.uni-marburg(dot)de
Projektbeschreibung
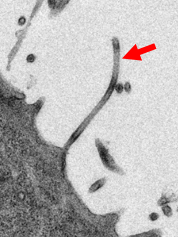
Elektronenmikroskopische Aufnahme von Ebolavirus infizierten Zellen. Die langgestreckten Viruspartikel werden von der Oberfläche der Zelle freigesetzt (roter Pfeil). ©Schauflinger
Das Ebolavirus (EBOV) verursacht schweres Fieber mit einer außerordentlich hohen Sterblichkeit. Das Matrixprotein VP40 von EBOV spielt eine Schlüsselrolle im viralen Replikationszyklus, der durch Homooligomerisierung des Proteins gesteuert wird. Die Dimerisierung von VP40 ist entscheidend für den Transport des Proteins zur Plasmamembran, wo die Dimere polymerisieren und zur Bildung von Filamenten führen, die die Knospung des Virus ermöglichen. Die VP40-Oktamerisierung führt zu einer Herunterregulierung der viralen RNA-Synthese. Aufgrund ihrer zentralen Rolle als Bausteine von Oligomeren höherer Ordnung stellen Dimere ein vielversprechendes Ziel für antivirale Medikamente dar. Mit Hilfe eines fragmentbasierten Ansatzes wurden kleine Moleküle identifiziert, die an VP40-Kristalle binden und zu Leitverbindungen weiterentwickelt werden sollen, um die VP40-Oligomerisierung und damit die Virusreplikation zu hemmen.
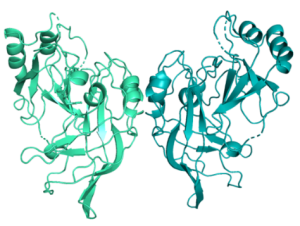
Hochauflösende Kristallstruktur des dimerischen Matrixproteins VP40 des Ebolavirus. ©Anke Werner
Wissenschaftliches Ziel:
Das Projekt zielt darauf ab, identifizierte Leitmoleküle zu antiviralen Medikamenten zu entwickeln, wobei strukturbasiertes Wirkstoffdesign – eine Kombination aus Proteinkristallographie und in silico-Methoden – sowie Zellkulturexperimente unter BSL4-Bedingungen zur Validierung eingesetzt werden.
DRUID-Kooperationspartner:
B1 Diederich/Kolb lab, A4 Heine/Reuter lab, D1 Steinmetzer lab, E3 Rahlfs/Przyborski lab
Literatur A1: 1. *Hartlieb et al. (2007) PNAS 104: 624-9 2. *Hartlieb et al., (2003) J. Biol. Chem. 278: 41830-6 3. *Hoenen et al. (2005) J Virol. 79: 1898-905 4. *Möller et al. 79, 14876-86 (2005) J Virol. 5. Hoenen et al. (2010) J Virol 84: 7053-63. 6. *Gomis-Ruth et al. (2003) Structure 11: 423-33.
A2Entwicklung von eIF4A-Inhibitoren als Kandidaten für eine klinische Anwendung sowie Charakterisierung von eIF4A-Varianten in Pathogenen
Arnold Grünweller

Prof. Dr. Arnold Grünweller
Philipps-Universität Marburg
Bau C
Marbacher Weg 6
35032 Marburg
Tel.: +49 (0)6421-2825849
Fax: +49 (0)6421-28 25854
E-Mail: arnold.gruenweller(at)staff.uni-marburg(dot)de
Projektbeschreibung
Die zelleigene RNA-Helikase eIF4A ist ein exzellentes Target für die Entwicklung antiviraler Breitbandmedikamente. Dieses Enzym wird für den Start der viralen Proteinsynthese von vielen Viren, insbesondere Coronaviren, benötigt und kann effizient und spezifisch durch die Stoffgruppe der Rocaglate gehemmt werden. In Projekt A2 möchten wir entsprechende Wirkstoffe für eine klinische Anwendung weiterentwickeln, indem wir unter anderem Rocaglate für eine lokale Applikation in den respiratorischen Trakt vernebeln und ein detailliertes Nebenwirkungsprofil erstellen. Zudem sollen neue eIF4A-Inhibitoren identifiziert und charakterisiert werden. Durch systematische Mutagenese bekannter Coronavirus-Sequenzen soll außerdem eine Vorhersage der Rocaglat-Sensitivität bei neu auftretenden Coronaviren ermöglicht werden. Schließlich soll die therapeutische Relevanz von Rocaglaten in verschiedenen DRUID-relevanten Pathogenen, die Varianten von eIF4A exprimieren, untersucht werden.
![]()

Festklemmen der RNA (RNA clamping) an der Oberfläche von eIF4A durch das Rocaglat Silvestrol sowie Nachweis der Hemmung der (corona)viralen Proteinsynthese durch das Rocaglat CR-31-B (-).
Wissenschaftliches Ziel:
Rocaglate sollen für eine klinische Anwendung weiterentwickelt und eine Vorhersage der Rocaglat-Sensitivität bei neu auftretenden Coronaviren und anderen DRUID-relevanten Pathogenen ermöglicht werden.
DRUID-Kooperationspartner:
B2 Ziebuhr lab, B4 Grevelding lab, B5 Schlitzer lab, C6 Häberlein lab, D1 Friebertshäuser/Steinmetzer lab, D3 van Zandbergen lab, D4 Hermosilla/Mazurek/Taubert lab, E4 Spengler lab, E6 Schiffmann lab
Literatur A2: 1.*Biedenkopf et al., (2017), Antiviral Res. 137: 76-81; 2. *Müller et al., (2018), Antiviral Res. 150:123-129; 3. *Elgner et al., (2018), Viruses. 10(4): 149; 4. *Glitscher et al., (2018), Viruses. 10(6): 301; 5. *Henß et al., (2018), Viruses. 10(11): 592; 6. *Müller et al., (2020), Antiviral Res. 175:104706; *7. *Müller et al., (2021), Antiviral Res. 186: 105012; 8. *Blum et al., (2020), J Cell Mol Med. 24(12): 6988-6999; [*eigene Publikationen].
A3Posttranslationale Proteinmodifikationen als Achillesferse pathogener RNA-Viren
Friedemann Weber

Prof. Dr. Friedemann Weber
Institut für Virologie
FB Veterinärmedizin
Justus-Liebig-Universität Gießen
Schubertstraße 81
35392 Gießen
Phone: +49 (0)641-99 38350
E-Mail: friedemann.weber(at)vetmed.uni-giessen(dot)de
Projektbeschreibung
Viren sind aufgrund ihres relativ kleinen Genoms stark von Funktionen der Wirtszellen abhängig. Viele dieser Funktionen werden durch zellkodierte, post-translationale Proteinmodifikationen reguliert, gegen die eine beträchtliche Anzahl pharmazeutischer Inhibitoren verfügbar ist.
Rift Valley Fever Virus (RVFV) ist ein in Afrika endemischer, Mücken-übertragener Zoonose-Erreger. In einem typischen RVFV-Ausbruch sterben Tausende Nutztiere und Hunderte Menschen. In der vorherigen Förderperiode haben wir mittels einer high-throughput Screening Plattform einen pro-viralen Wirtszellfaktor für RVFV identifiziert, der an posttranslationale Proteinmodifikationen bindet. Inhibition dieses Faktors führte in human-Organoiden zu einer deutlichen Reduktion der viralen RNA-Synthese und der Virusproduktion. Zudem ergaben Proteom-Analysen, dass ein RVFV-Protein posttranslational so modifiziert wird, dass der neue Wirtsfaktor binden kann, und dass die Mutation dieser Modifikationsstelle die virale RNA-Synthese reduziert.
Wissenschaftliches Ziel:
Das geplante Projekt zielt darauf ab, essentielle Interaktionen von VP40 mit sich selbst durch synthetische Peptide oder kleine Moleküle zu hemmen und dadurch die Virusvermehrung zu blockieren. Eine der wesentlichen Herausforderungen dieses Projektes ist die Identifizierung von zellgängigen nicht (zyto)toxischen Inhibitoren.
DRUID-Kooperationspartner:
A1 Becker, A2 Grünweller, B2 Ziebuhr, C1 Bender/Hildt, D1 Friebertshäuser/Steinmetzer, E3 Rahlfs/ Przyborski, E4 Spengler, E6 Schiffmann, E7P Krijnse Locker
Literatur A3: 1. Wuerth & Weber (2016) Viruses 8, 174*. 2. Barr, Weber, Schmaljohn (2020) Fields Virology, vol 1, p 706-749*
A4Leitstruktursuche zur Inhibierung des Chaperons IpgC aus Shigella sp.
Andreas Heine, Klaus Reuter

Prof. Dr. Andreas Heine
Institut für Pharmazeutische Chemie
Marbacher Weg 6
35032 Marburg
Tel.: +49 (0)6421-28 21313
Fax: +49 (0)6421-28 28994
E-Mail: heinea(at)staff.uni-marburg(dot)de

Prof. Dr. Klaus Reuter
Institut für Pharmazeutische Chemie
Philipps-Universität Marburg
Marbacher Weg 6
35032 Marburg
Tel.: +49 (0)6421-28 25845
Fax: +49 (0)6421-28 28994
E-Mail: reuterk(at)staff.uni-marburg(dot)de
Projektbeschreibung
Bakterien der Gattung Shigella dringen in die Epithelzellen des Colons ein und verursachen eine entzündliche Erkrankung des Dickdarms. Letztere ist als „Bakterienruhr“ bekannt und fordert v.a. in Entwicklungsländern zahlreiche Todesopfer. Das Shigella-spezifische Chaperon IpgC ist für die Pathogenität des Erregers essentiell und tritt mit zahlreichen weiteren Pathogenitätsfaktoren in Wechselwirkung. In Abwesenheit eines „Substrat-Proteins“ liegt IpgC als Homodimer vor, was für die Aufrechterhaltung seiner Stabilität erforderlich ist. Wir nutzen IpgC als Zielprotein für die strukturbasierte Entwicklung von Wirkstoffen gegen Bakterienruhr, indem wir durch Kleinmoleküle die Homodimerbildung und/oder sein Binden an „Substrat-Proteine“ verhindern. Grundlage dafür ist ein von uns etabliertes Protokoll, welches reproduzierbar hervorragend streuende Kristalle von IpgC liefert. Ein bereits durchgeführtes Fragment-Screening führte zu mehreren Treffern, von denen einige mittlerweile zu größeren „Bindern“ erweitert werden konnten. Um deren Effekt auf die Homodimerbildung, auf die Wechselwirkung mit Substraten und auf die Stabilität des Proteins zu untersuchen, setzen wir neben der Proteinkristallographie Methoden wie „Mikroskalierte Thermophorese“, „Isothermische Titrationskalorimetrie“ sowie ein „Thermal Shift“-Assay ein.

Kristallstruktur des IpgC-Homodimers ©Klaus Reuter
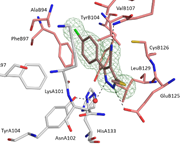
An IpgC-gebundenes „erweitertes Fragment“ ©Marina Gardonyi
Wissenschaftliches Ziel:
Hauptziel ist neben zusätzlicher Strukturinformation über IpgC die Optimierung der bisher erhaltenen Verbindungen sowie ihr Einsatz als Leitstrukturen bei der Anti-Shigellose-Wirkstoffentwicklung.
DRUID-Kooperationspartner:
A1 Stephan Becker, B1 Wibke Diederich / Peter Kolb, B7 Franco Falcone, D1 Eva Friebertshäuser / Torsten Steinmetzer
Literatur A4: [1] Agerberth et al. (2005) World Health Organ [2] Williams & Berkley (2018) Paediatr Int Child Health 38:50-65. [3] Sansonetti (2001) Am J Physiol Liver Physiol 280:319-323. [4] Parsot et al. (2003) Curr Opin Microbiol 6:7-14. [5] Lunelli et al. (2009) Proc Natl Acad Sci USA 106:9661-9666.
A6Die hoch divergente Aktin-Superfamilie als Wirkstoffziel bei der Übertragung von Malariaparasiten
Ross Douglas

Dr. Ross Douglas
Biomedical Research Center Seltersberg (BFS)
Molecular Infections Biology
Justus Liebig Universität Giessen
Schubertstrasse 81
35392 Gießen
Tel.: +49 (0)641-99 39145
Fax: +49 (0)641-99 39129
E-Mail: ross.g.douglas(at)ernaehrung.uni-giessen(dot)de
Projektbeschreibung
Malaria ist nach wie vor eine der verheerendsten Krankheiten und wird durch einzellige Parasiten namens Plasmodium verursacht. Diese Parasiten werden durch Anopheles-Moskitos von Mensch zu Mensch übertragen. Der Parasit benötigt eine Reihe verschiedener Proteine, die ihm die Übertragung auf den Moskito-Vektor ermöglichen, darunter Mitglieder des stark divergierenden Aktin-Zytoskeletts und seiner Regulatoren. Das Zytoskelett des Parasiten hat einzigartige Eigenschaften, um die Übertragung zu ermöglichen. Da es für verschiedene Prozesse des Parasiten in unterschiedlichen Stadien des Lebenszyklus unerlässlich ist, enthält es vielversprechende Ziele für neuartige Malariatherapien. Wir nutzen Ansätze zur Target-Validierung, um neue Targets zu identifizieren und zu charakterisieren, die die Übertragung blockieren und zur Kontrolle der Krankheitsausbreitung eingesetzt werden könnten.
Der Ansatz der Target-Validierung hat neue Targets identifiziert, die die Übertragung blockieren.
Wissenschaftliches Ziel:
Unser Ziel ist es, die identifizierten Proteine mit in vitro und in vivo Methoden zu charakterisieren, um neue Wirkstoffe zu identifizieren, die selektiv auf diese Proteine abzielen und somit als Kandidaten für Medikamente zur Blockierung der Übertragung dienen.
DRUID-Kooperationspartner:
A7 Przyborski, B1 Diederich/Kolb, B7 P Falcone, E3 Rahlfs/Przyborski
Literatur A6: [1] Douglas et al. (2018) PLOS Bio e2005345; [2] Douglas et al. (2018) Malaria J 17:3191898-905; [3] Douglas et al. (2015) Trends Parasitol 31(8):357-362.
A7Plasmodium falciparum Chaperone, Co-Chaperone und deren Protein-Protein-Interaktionen als Ziele für neue Interventionsstrategien
Jude Przyborski

Prof. Dr. Jude Przyborski
Interdisziplinäres Forschungszentrum (iFZ)
Justus-Liebig-Universität Gießen
Heinrich-Buff-Ring 26-32
35392 Gießen
Tel.: +49 (0)641-99 39114
E-Mail: jude.przyborski(at)ernaehrung.uni-giessen(dot)de
Projektbeschreibung
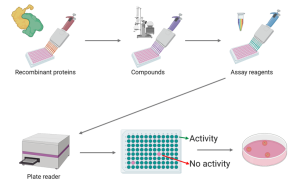
Übersicht der Assay-Entwicklung
Malariaparasiten dringen in reife menschliche rote Blutkörperchen (RBC) ein und leben darin. Um sein Überleben zu sichern, baut der Parasit die von ihm gewählte Wirtszelle zu seinem eigenen Vorteil um. Infizierte rote Blutkörperchen werden klebrig und haften an der Innenwand kleiner Blutgefäße, außerdem überziehen sie sich mit Proteinen, die es ihnen ermöglichen, für das Immunsystem unsichtbar zu werden. Dies führt leider zur Krankheit des Patienten und schließlich zum Tod. Wir haben kürzlich eine Reihe wichtiger molekularer Akteure identifiziert, die für diesen Umbauprozess wesentlich sind, darunter Mitglieder der so genannten HSP70- und HSP40-Familien. Das Ziel dieses Projekts ist es, die Funktion von HSP40 und HSP70 zu blockieren. Wenn uns dies gelingt, werden die Parasiten wahrscheinlich aus dem Blutkreislauf entfernt, wodurch die Schwere der Krankheit gelindert wird. Zu diesem Zweck werden wir eine Reihe von Assays zur Messung der Aktivität von HSP40/HSP70 entwickeln und auf dieser Grundlage nach Verbindungen suchen, die diese Wechselwirkung verringern. Vielversprechende Verbindungen werden dann direkt an Parasiten auf ihre Fähigkeit getestet, die Veränderung der Wirtszellen zu verringern.
Literatur A7: 1. Diehl et al. (2021) PLoS Pathogens 17:e1009969 2. Zhang et al. (2017) Sci Rep 7: 42188 3. Charnaud et al. (2017) PLoS One 12: e0181656 4. Külzer et al. (2012) Cell Micro 14: 1784-95 5. Külzer et al. (2010) Cell Micro 12: 1398-1420